Titan Workflow Series¶
The Titan Workflow Series is a collection of WDL workflows developed for performing genomic characterization and genomic epidemiology of viral samples to support public health decision-making. As of today (May 4th, 2021) these workflows are specific to SARS-CoV-2 amplicon read data, but work is underway to allow for the analysis of other viral pathogens of concern.
Titan Workflows for Genomic Characterization¶
Genomic characterization, i.e. generating consensus assemblies (FASTA format) from next-generation sequencing (NGS) read data (FASTQ format) to assign samples with relevant nomenclature designation (e.g. PANGO lineage and NextClade clades) is an increasingly critical function to public health laboratories around the world.
The Titan Series includes four separate WDL workflows (Titan_Illumina_PE, Titan_Illumina_SE, Titan_ClearLabs, and Titan_ONT) that process NGS read data from four different sequencing approaches: Illumina paired-end, Illumina single-end, Clear Labs, and Oxford Nanopore Technology (ONT)) to generate consensus assemblies, produce relevant quality-control metrics for both the input read data and the generated assembly, and assign samples with a lineage and clade designation using Pangolin and NextClade, respectively.
All four Titan workflows for genomic characterization will generate a viral assembly by mapping input read data to a reference genome, removing primer reads from that alignment, and then calling the consensus assembly based on the primer-trimmed alignment. These consensus assemblies are then fed into the Pangolin and NextClade CLI tools for lineage and clade assignments.
The major difference between each of these Titan workflows is in how the read mapping, primer trimming, and consensus genome calling is performed. More information on the technical details of these processes and information on how to utilize and apply these workflows for public health investigations is available below.
A series of introductory training videos that provide conceptual overviews of methodologies and walkthrough tutorials on how to utilize these Titan workflows through Terra are available on the Theiagen Genomics YouTube page:
Titan_Illumina_PE¶
The Titan_Illumina_PE workflow was written to process Illumina paired-end (PE) read data. Input reads are assumed to be the product of sequencing tiled PCR-amplicons designed for the SARS-CoV-2 genome. The most common read data analyzed by the Titan_Illumina_PE workflow are generated with the Artic V3 protocol. Alternative primer schemes such as the Qiaseq Primer Panel, however, can also be analysed with this workflow. The primer sequence coordinates of the PCR scheme utilized must be provided along with the raw paired-end Illumina read data in BED and FASTQ file formats, respectively.
Note
By default, this workflow will assume that input reads were generated using a 300-cycle kit (i.e. 2 x 150 bp reads). Modifications to the optional parameter for trimmomatic_minlen may be required to accommodate for shorter read data, such as 2 x 75bp reads generated using a 150-cycle kit.
Upon initiating a Titan_Illumina_PE job, the input primer scheme coordinates and raw paired-end Illumina read data provided for each sample will be processed to perform consensus genome assembly, infer the quality of both raw read data and the generated consensus genome, and assign samples SARS-CoV-2 lineage and clade types as outlined in the Titan_Illumina_PE data workflow below.
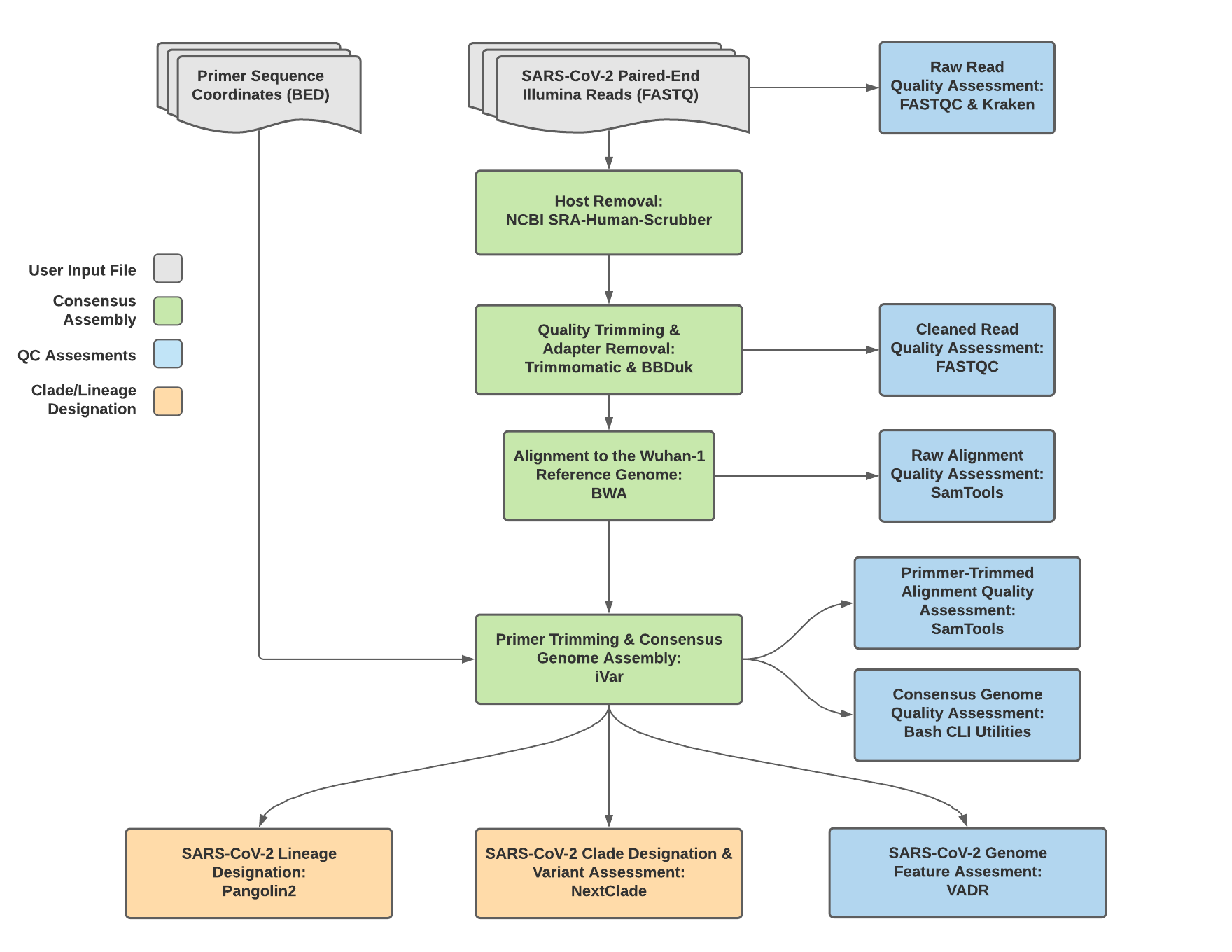
Titan_Illumina_PE v1.4.4 Data Workflow¶
Consensus genome assembly with the Titan_Illumina_PE workflow is performed by first de-hosting read data with the NCBI SRA-Human-Scrubber tool then trimming low-quality reads with Trimmomatic and removing adapter sequences with BBDuk. These cleaned read data are then aligned to the Wuhan-1 reference genome with BWA to generate a Binary Alignment Mapping (BAM) file. Primer sequences are then removed from the BAM file using the iVar Trim sub-command. The iVar consensus sub-command is then utilized to generate a consensus assembly in FASTA format. This assembly is then used to assign lineage and clade designations with Pangolin and NextClade. NCBI’S VADR tool is also employed to screen for potentially errant features (e.g. erroneous frame-shift mutations) in the consensus assembly.
More information on required user inputs, optional user inputs, default tool parameters and the outputs generated by Titan_Illumina_PE are outlined below.
Required User Inputs¶
Download CSV: Titan_Illumina_PE_required_inputs.csv
Task |
Input Variable |
Data Type |
Description |
|---|---|---|---|
titan_illumina_pe |
primer_bed |
File |
Primer sequence coordinates of the PCR scheme utilized in BED file format |
titan_illumina_pe |
read1_raw |
File |
Forward Illumina read in FASTQ file format |
titan_illumina_pe |
read2_raw |
File |
Reverse Illumina read in FASTQ file format |
titan_illumina_pe |
samplename |
String |
Name of the sample being analyzed |
Optional User Inputs¶
Download CSV: Titan_Illumina_PE_optional_inputs.csv
Task |
Variable Name |
Data Type |
Description |
Default |
|---|---|---|---|---|
bedtools_cov |
primer_bed |
String |
Path to the primer sequence coordinates of the PCR scheme utilized in BED file format |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019_amplicon.bed |
bedtools_cov |
fail_threshold |
String |
Minimum coverage threshold to determin amplicon sequencing failture |
20x |
bwa |
reference_genome |
String |
Path to the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta |
bwa |
cpus |
Int |
CPU resources allocated to the BWA task runtime environment |
6 |
consensus |
ref_gff |
String |
Path to the general feature format of the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/reference/GCF_009858895.2_ASM985889v3_genomic.gff |
consensus |
ref_genome |
String |
Path to the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta |
consensus |
min_qual |
Int |
Minimum quality threshold for sliding window to pass for iVar consensus |
20 |
consensus |
min_freq |
Float |
Minimum frequency threshold(0 - 1) to call variants for iVar consensus |
0.6 |
consensus |
min_depth |
Int |
Minimum read depth to call variants for iVar consensus |
10 |
consensus |
min_bq |
Int |
Minimum mapping quality for an alignment to be used for SAMtools mpileup before running iVar consensus |
0 |
consensus |
max_depth |
Int |
Maximum reads read at a position per input file for SAMtools mpileup before running iVar consensus |
600000 |
consensus |
disable_baq |
Boolean |
Disable read-pair overlap detection for SAMtools mpileup before running iVar consensus |
TRUE |
consensus |
count_orphans |
Boolean |
Do not skip anomalous read pairs in variant calling for SAMtools mpileup before running iVar consensus |
TRUE |
consensus |
char_unknown |
String |
Character to print in regions with less than minimum coverage for iVar consensus |
N |
nextclade_one_sample |
root_sequence |
File |
Custom reference sequence file for NextClade |
None |
nextclade_one_sample |
qc_config_json |
File |
Custom QC configuraiton file for NextClade |
None |
nextclade_one_sample |
pcr_primers_csv |
File |
Custom PCR primers file for NextClade |
None |
nextclade_one_sample |
gene_annotations_json |
File |
Custom gene annotation file for NextClade |
None |
nextclade_one_sample |
docker |
String |
Docker tag used for running NextClade |
neherlab/nextclade:0.14.2 |
nextclade_one_sample |
auspice_reference_tree_json |
File |
Custom reference tree file for NextClade |
None |
pangolin3 |
inference_engine |
String |
pangolin inference engine for lineage designations (usher or pangolarn) |
usher |
pangolin3 |
min_length |
Int |
Minimum query length allowed for pangolin to attempt assignment |
10000 |
pangolin3 |
max_ambig |
Float |
Maximum proportion of Ns allowed for pangolin to attempt assignment |
0.5 |
primer_trim |
keep_noprimer_reads |
Boolean |
Include reads with no primers for iVar trim |
True |
read_QC_trim |
trimmomatic_window_size |
Int |
Specifies the number of bases to average across for Trimmomatic |
4 |
read_QC_trim |
trimmomatic_quality_trim_score |
Int |
Specifies the average quality required for Trimmomatic |
30 |
read_QC_trim |
trimmomatic_minlen |
Int |
Specifies the minimum length of reads to be kept for Trimmomatic |
75 |
titan_illumina_pe |
seq_method |
String |
Description of the sequencing methodology used to generate the input read data |
Illumina paired-end |
titan_illumina_pe |
pangolin_docker_image |
String |
Docker tag used for running Pangolin |
staphb/pangolin:2.4.2-pangolearn-2021-05-19 |
vadr |
docker |
String |
Docker tag used for running VADR |
staphb/vadr:1.2.1 |
vadr |
maxlen |
Int |
Maximum length for the fasta-trim-terminal-ambigs.pl VADR script |
30000 |
vadr |
minlen |
Int |
Minimum length subsequence to possibly replace Ns for the fasta-trim-terminal-ambigs.pl VADR script |
50 |
vadr |
vadr_opts |
String |
Options for the v-annotate.pl VADR script |
–glsearch -s -r –nomisc –mkey sarscov2 –alt_fail lowscore,fstukcnf,insertnn,deletinn –mdir /opt/vadr/vadr-models/ |
vadr |
skip_length |
Int |
Minimum assembly length (unambiguous) to run vadr |
10000 |
variant_call |
ref_gff |
String |
Path to the general feature format of the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/reference/GCF_009858895.2_ASM985889v3_genomic.gff |
variant_call |
ref_genome |
String |
Path to the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta |
variant_call |
min_qual |
Int |
Minimum quality threshold for sliding window to pass for iVar variants |
20 |
variant_call |
min_freq |
Float |
Minimum frequency threshold(0 - 1) to call variants for iVar variants |
0.6 |
variant_call |
min_depth |
Int |
Minimum read depth to call variants for iVar variants |
10 |
variant_call |
min_bq |
Int |
Minimum mapping quality for an alignment to be used for SAMtools mpileup before running iVar variants |
0 |
variant_call |
max_depth |
Int |
Maximum reads read at a position per input file for SAMtools mpileup before running iVar variants |
600000 |
variant_call |
disable_baq |
Boolean |
Disable read-pair overlap detection for SAMtools mpileup before running iVar variants |
TRUE |
variant_call |
count_orphans |
Boolean |
Do not skip anomalous read pairs in variant calling for SAMtools mpileup before running iVar variants |
TRUE |
version_capture |
timezone |
String |
User time zone in valid Unix TZ string (e.g. America/New_York) |
None |
Outputs¶
Download CSV: Titan_Illumina_PE_default_outputs.csv
Output Name |
Data Type |
Description |
|---|---|---|
aligned_bai |
File |
Index companion file to the bam file generated during the consensus assembly process |
aligned_bam |
File |
Primer-trimmed BAM file; generated during conensus assembly process |
assembly_fasta |
File |
Consensus genome assembly |
assembly_length_unambiguous |
Int |
Number of unambiguous basecalls within the SC2 consensus assembly |
assembly_mean_coverage |
Float |
Mean sequencing depth throughout the conesnsus assembly generated after performing primer trimming–calculated using the SAMtools coverage command |
assembly_method |
String |
Method employed to generate consensus assembly |
auspice_json |
File |
Auspice-compatable JSON output generated from NextClade analysis that includes the NextClade default samples for clade-typing and the single sample placed on this tree |
bbduk_docker |
String |
Docker image used to run BBDuk |
bwa_version |
String |
Version of BWA used to map read data to the reference genome |
consensus_flagstat |
File |
Output from the SAMtools flagstat command to assess quality of the alignment file (BAM) |
consensus_stats |
File |
Output from the SAMtools stats command to assess quality of the alignment file (BAM) |
dehosted_read1 |
File |
Dehosted forward reads; suggested read file for SRA submission |
dehosted_read2 |
File |
Dehosted reverse reads; suggested read file for SRA submission |
fastqc_clean_pairs |
String |
Number of paired reads after SeqyClean filtering as determined by FastQC |
fastqc_clean1 |
Int |
Number of forward reads after seqyclean filtering as determined by FastQC |
fastqc_clean2 |
Int |
Number of reverse reads after seqyclean filtering as determined by FastQC |
fastqc_raw_pairs |
String |
Number of paired reads identified in the input fastq files as determined by FastQC |
fastqc_raw1 |
Int |
Number of forward reads identified in the input fastq files as determined by FastQC |
fastqc_raw2 |
Int |
Number of reverse reads identified in the input fastq files as determined by FastQC |
fastqc_version |
String |
Version of the FastQC software used for read QC analysis |
ivar_tsv |
File |
Variant descriptor file generated by iVar variants |
ivar_variant_version |
String |
Version of iVar for running the iVar variants command |
ivar_version_consensus |
String |
Version of iVar for running the iVar consensus command |
ivar_version_primtrim |
String |
Version of iVar for running the iVar trim command |
kraken_human |
Float |
Percent of human read data detected using the Kraken2 software |
kraken_human_dehosted |
Float |
Percent of human read data detected using the Kraken2 software after host removal |
kraken_report |
File |
Full Kraken report |
kraken_report_dehosted |
File |
Full Kraken report after host removal |
kraken_sc2 |
Float |
Percent of SARS-CoV-2 read data detected using the Kraken2 software |
kraken_sc2_dehosted |
Float |
Percent of SARS-CoV-2 read data detected using the Kraken2 software after host removal |
kraken_version |
String |
Version of Kraken software used |
meanbaseq_trim |
Float |
Mean quality of the nucleotide basecalls aligned to the reference genome after primer trimming |
meanmapq_trim |
Float |
Mean quality of the mapped reads to the reference genome after primer trimming |
nextclade_aa_dels |
String |
Amino-acid deletions as detected by NextClade |
nextclade_aa_subs |
String |
Amino-acid substitutions as detected by NextClade |
nextclade_clade |
String |
NextClade clade designation |
nextclade_json |
File |
NexClade output in JSON file format |
nextclade_tsv |
File |
NextClade output in TSV file format |
nextclade_version |
String |
Version of NextClade software used |
number_Degenerate |
Int |
Number of degenerate basecalls within the consensus assembly |
number_N |
Int |
Number of fully ambiguous basecalls within the consensus assembly |
number_Total |
Int |
Total number of nucleotides within the consensus assembly |
pango_lineage |
String |
Pango lineage as detremined by Pangolin |
pango_lineage_report |
File |
Full Pango lineage report generated by Pangolin |
pangolin_conflicts |
String |
Number of lineage conflicts as deteremed by Pangolin |
pangolin_docker |
String |
Docker image used to run Pangolin |
pangolin_notes |
String |
Lineage notes as deteremined by Pangolin |
pangolin_version |
String |
Pangolin and PangoLEARN versions used |
percent_reference_coverage |
Float |
Percent coverage of the reference genome after performing primer trimming; calculated as assembly_length_unambiguous / length of reference genome (SC2: 29,903) x 100 |
primer_trimmed_read_percent |
Float |
Percent of read data with primers trimmed as deteremined by iVar trim |
read1_clean |
File |
Forward read file after quality trimming and adapter removal |
read2_clean |
File |
Reverse read file after quality trimming and adapter removal |
samtools_version |
String |
Version of SAMtools used to sort and index the alignment file |
samtools_version_consensus |
String |
Version of SAMtools used to create the pileup before running iVar consensus |
samtools_version_primtrim |
String |
Version of SAMtools used to create the pileup before running iVar trim |
samtools_version_stats |
String |
Version of SAMtools used to assess quality of read mapping |
seq_platform |
String |
Description of the sequencing methodology used to generate the input read data |
titan_illumina_pe_analysis_date |
String |
Date of analysis |
titan_illumina_pe_version |
String |
Version of the Public Health Viral Genomics (PHVG) repository used |
trimmomatic_version |
String |
Version of Trimmomatic used |
vadr_alerts_list |
File |
File containing all of the fatal alerts as determined by VADR |
vadr_docker |
String |
Docker image used to run VADR |
vadr_num_alerts |
String |
Number of fatal alerts as determined by VADR |
Titan_Illumina_SE¶
The Titan_Illumina_SE workflow was written to process Illumina single-end (SE) read data. Input reads are assumed to be the product of sequencing tiled PCR-amplicons designed for the SARS-CoV-2 genome. The most common read data analyzed by the Titan_Illumina_SE workflow are generated with the Artic V3 protocol. Alternative primer schemes such as the Qiaseq Primer Panel, however, can also be analysed with this workflow. The primer sequence coordinates of the PCR scheme utilized must be provided along with the raw paired-end Illumina read data in BED and FASTQ file formats, respectively.
Note
By default, this workflow will assume that input reads were generated using a 35-cycle kit (i.e. 1 x 35 bp reads). Modifications to the optional parameter for trimmomatic_minlen may be required to accommodate for longer read data.
Upon initiating a Titan_Illumina_SE job, the input primer scheme coordinates and raw paired-end Illumina read data provided for each sample will be processed to perform consensus genome assembly, infer the quality of both raw read data and the generated consensus genome, and assign samples SARS-CoV-2 lineage and clade types as outlined in the Titan_Illumina_PE data workflow below.
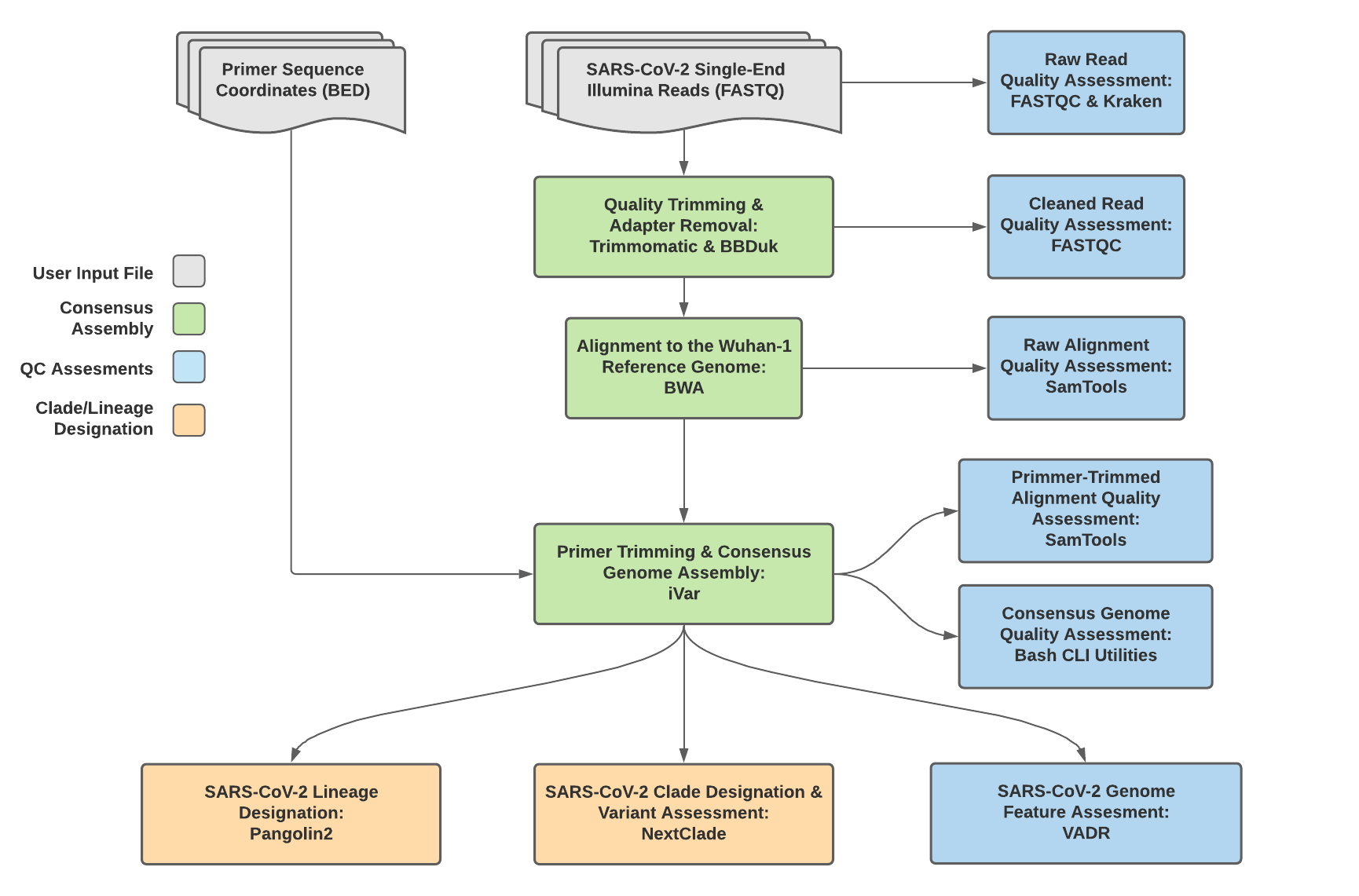
Titan_Illumina_SE v1.4.4 Data Workflow¶
Consensus genome assembly with the Titan_Illumina_SE workflow is performed by first trimming low-quality reads with Trimmomatic and removing adapter sequences with BBDuk. These cleaned read data are then aligned to the Wuhan-1 reference genome with BWA to generate a Binary Alignment Mapping (BAM) file. Primer sequences are then removed from the BAM file using the iVar Trim sub-command. The iVar consensus sub-command is then utilized to generate a consensus assembly in FASTA format. This assembly is then used to assign lineage and clade designations with Pangolin and NextClade. NCBI’S VADR tool is also employed to screen for potentially errant features (e.g. erroneous frame-shift mutations) in the consensus assembly.
More information on required user inputs, optional user inputs, default tool parameters and the outputs generated by Titan_Illumina_SE are outlined below.
Required User Inputs¶
Download CSV: Titan_Illumina_SE_required_inputs.csv
Task |
Input Variable |
Data Type |
Description |
|---|---|---|---|
titan_illumina_pe |
primer_bed |
File |
Primer sequence coordinates of the PCR scheme utilized in BED file format |
titan_illumina_pe |
read1_raw |
File |
Single-end Illumina read in FASTQ file format |
titan_illumina_pe |
samplename |
String |
Name of the sample being analyzed |
Optional User Inputs¶
Download CSV: Titan_Illumina_SE_optional_inputs.csv
Task |
Variable Name |
Data Type |
Description |
Default |
|---|---|---|---|---|
bedtools_cov |
primer_bed |
String |
Path to the primer sequence coordinates of the PCR scheme utilized in BED file format |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019_amplicon.bed |
bedtools_cov |
fail_threshold |
String |
Minimum coverage threshold to determin amplicon sequencing failture |
20x |
bwa |
reference_genome |
String |
Path to the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta |
bwa |
cpus |
Int |
CPU resources allocated to the BWA task runtime environment |
6 |
bwa |
read2 |
File |
Optional input file for the bwa task that is not applicable to this workflow |
None |
consensus |
ref_gff |
String |
Path to the general feature format of the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/reference/GCF_009858895.2_ASM985889v3_genomic.gff |
consensus |
ref_genome |
String |
Path to the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta |
consensus |
min_qual |
Int |
Minimum quality threshold for sliding window to pass for iVar consensus |
20 |
consensus |
min_freq |
Float |
Minimum frequency threshold(0 - 1) to call variants for iVar consensus |
0.6 |
consensus |
min_depth |
Int |
Minimum read depth to call variants for iVar consensus |
10 |
consensus |
min_bq |
Int |
Minimum mapping quality for an alignment to be used for SAMtools mpileup before running iVar consensus |
0 |
consensus |
max_depth |
Int |
Maximum reads read at a position per input file for SAMtools mpileup before running iVar consensus |
600000 |
consensus |
disable_baq |
Boolean |
Disable read-pair overlap detection for SAMtools mpileup before running iVar consensus |
TRUE |
consensus |
count_orphans |
Boolean |
Do not skip anomalous read pairs in variant calling for SAMtools mpileup before running iVar consensus |
TRUE |
consensus |
char_unknown |
String |
Character to print in regions with less than minimum coverage for iVar consensus |
N |
nextclade_one_sample |
root_sequence |
File |
Custom reference sequence file for NextClade |
None |
nextclade_one_sample |
qc_config_json |
File |
Custom QC configuraiton file for NextClade |
None |
nextclade_one_sample |
pcr_primers_csv |
File |
Custom PCR primers file for NextClade |
None |
nextclade_one_sample |
gene_annotations_json |
File |
Custom gene annotation file for NextClade |
None |
nextclade_one_sample |
docker |
String |
Docker tag used for running NextClade |
neherlab/nextclade:0.14.2 |
nextclade_one_sample |
auspice_reference_tree_json |
File |
Custom reference tree file for NextClade |
None |
pangolin3 |
inference_engine |
String |
pangolin inference engine for lineage designations (usher or pangolarn) |
usher |
pangolin3 |
min_length |
Int |
Minimum query length allowed for pangolin to attempt assignment |
10000 |
pangolin3 |
max_ambig |
Float |
Maximum proportion of Ns allowed for pangolin to attempt assignment |
0.5 |
primer_trim |
keep_noprimer_reads |
Boolean |
Include reads with no primers for iVar trim |
True |
read_QC_trim |
trimmomatic_window_size |
Int |
Specifies the number of bases to average across for Trimmomatic |
4 |
read_QC_trim |
trimmomatic_quality_trim_score |
Int |
Specifies the average quality required for Trimmomatic |
30 |
read_QC_trim |
trimmomatic_minlen |
Int |
Specifies the minimum length of reads to be kept for Trimmomatic |
25 |
titan_illumina_pe |
seq_method |
String |
Description of the sequencing methodology used to generate the input read data |
Illumina paired-end |
titan_illumina_pe |
pangolin_docker_image |
String |
Docker tag used for running Pangolin |
staphb/pangolin:2.4.2-pangolearn-2021-05-19 |
vadr |
docker |
String |
Docker tag used for running VADR |
staphb/vadr:1.2.1 |
vadr |
maxlen |
Int |
Maximum length for the fasta-trim-terminal-ambigs.pl VADR script |
30000 |
vadr |
minlen |
Int |
Minimum length subsequence to possibly replace Ns for the fasta-trim-terminal-ambigs.pl VADR script |
50 |
vadr |
vadr_opts |
String |
Options for the v-annotate.pl VADR script |
–glsearch -s -r –nomisc –mkey sarscov2 –alt_fail lowscore,fstukcnf,insertnn,deletinn –mdir /opt/vadr/vadr-models/ |
vadr |
skip_length |
Int |
Minimum assembly length (unambiguous) to run vadr |
10000 |
variant_call |
ref_gff |
String |
Path to the general feature format of the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/reference/GCF_009858895.2_ASM985889v3_genomic.gff |
variant_call |
ref_genome |
String |
Path to the reference genome within the staphb/ivar:1.2.2_artic20200528 Docker container |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta |
variant_call |
min_qual |
Int |
Minimum quality threshold for sliding window to pass for iVar variants |
20 |
variant_call |
min_freq |
Float |
Minimum frequency threshold(0 - 1) to call variants for iVar variants |
0.6 |
variant_call |
min_depth |
Int |
Minimum read depth to call variants for iVar variants |
10 |
variant_call |
min_bq |
Int |
Minimum mapping quality for an alignment to be used for SAMtools mpileup before running iVar variants |
0 |
variant_call |
max_depth |
Int |
Maximum reads read at a position per input file for SAMtools mpileup before running iVar variants |
600000 |
variant_call |
disable_baq |
Boolean |
Disable read-pair overlap detection for SAMtools mpileup before running iVar variants |
TRUE |
variant_call |
count_orphans |
Boolean |
Do not skip anomalous read pairs in variant calling for SAMtools mpileup before running iVar variants |
TRUE |
version_capture |
timezone |
String |
User time zone in valid Unix TZ string (e.g. America/New_York) |
None |
Outputs¶
Download CSV: Titan_Illumina_SE_default_outputs.csv
Output Name |
Data Type |
Description |
|---|---|---|
aligned_bai |
File |
Index companion file to the bam file generated during the consensus assembly process |
aligned_bam |
File |
Primer-trimmed BAM file; generated during conensus assembly process |
assembly_fasta |
File |
Consensus genome assembly |
assembly_length_unambiguous |
Int |
Number of unambiguous basecalls within the SC2 consensus assembly |
assembly_mean_coverage |
Float |
Mean sequencing depth throughout the conesnsus assembly generated after performing primer trimming–calculated using the SAMtools coverage command |
assembly_method |
String |
Method employed to generate consensus assembly |
auspice_json |
File |
Auspice-compatable JSON output generated from NextClade analysis that includes the NextClade default samples for clade-typing and the single sample placed on this tree |
bbduk_docker |
String |
Docker image used to run BBDuk |
bwa_version |
String |
Version of BWA used to map read data to the reference genome |
consensus_flagstat |
File |
Output from the SAMtools flagstat command to assess quality of the alignment file (BAM) |
consensus_stats |
File |
Output from the SAMtools stats command to assess quality of the alignment file (BAM) |
fastqc_clean |
Int |
Number of reads after SeqyClean filtering as determined by FastQC |
fastqc_raw |
Int |
Number of reads after seqyclean filtering as determined by FastQC |
fastqc_version |
String |
Version of the FastQC software used for read QC analysis |
ivar_tsv |
File |
Variant descriptor file generated by iVar variants |
ivar_variant_version |
String |
Version of iVar for running the iVar variants command |
ivar_version_consensus |
String |
Version of iVar for running the iVar consensus command |
ivar_version_primtrim |
String |
Version of iVar for running the iVar trim command |
kraken_human |
Float |
Percent of human read data detected using the Kraken2 software |
kraken_report |
String |
Full Kraken report |
kraken_sc2 |
Float |
Percent of SARS-CoV-2 read data detected using the Kraken2 software |
kraken_version |
String |
Version of Kraken software used |
meanbaseq_trim |
Float |
Mean quality of the nucleotide basecalls aligned to the reference genome after primer trimming |
meanmapq_trim |
Float |
Mean quality of the mapped reads to the reference genome after primer trimming |
nextclade_aa_dels |
String |
Amino-acid deletions as detected by NextClade |
nextclade_aa_subs |
String |
Amino-acid substitutions as detected by NextClade |
nextclade_clade |
String |
NextClade clade designation |
nextclade_json |
File |
NexClade output in JSON file format |
nextclade_tsv |
File |
NextClade output in TSV file format |
nextclade_version |
String |
Version of NextClade software used |
number_Degenerate |
Int |
Number of degenerate basecalls within the consensus assembly |
number_N |
Int |
Number of fully ambiguous basecalls within the consensus assembly |
number_Total |
Int |
Total number of nucleotides within the consensus assembly |
pango_lineage |
String |
Pango lineage as detremined by Pangolin |
pango_lineage_report |
File |
Full Pango lineage report generated by Pangolin |
pangolin_conflicts |
String |
Number of lineage conflicts as deteremed by Pangolin |
pangolin_docker |
String |
Docker image used to run Pangolin |
pangolin_notes |
String |
Lineage notes as deteremined by Pangolin |
pangolin_version |
String |
Pangolin and PangoLEARN versions used |
percent_reference_coverage |
Float |
Percent coverage of the reference genome after performing primer trimming; calculated as assembly_length_unambiguous / length of reference genome (SC2: 29,903) x 100 |
primer_trimmed_read_percent |
Float |
Percent of read data with primers trimmed as deteremined by iVar trim |
read1_clean |
File |
Forward read file after quality trimming and adapter removal |
samtools_version |
String |
Version of SAMtools used to sort and index the alignment file |
samtools_version_consensus |
String |
Version of SAMtools used to create the pileup before running iVar consensus |
samtools_version_primtrim |
String |
Version of SAMtools used to create the pileup before running iVar trim |
samtools_version_stats |
String |
Version of SAMtools used to assess quality of read mapping |
seq_platform |
String |
Description of the sequencing methodology used to generate the input read data |
titan_illumina_se_analysis_date |
String |
Date of analysis |
titan_illumina_se_version |
String |
Version of the Public Health Viral Genomics (PHVG) repository used |
trimmomatic_version |
String |
Version of Trimmomatic used |
vadr_alerts_list |
File |
File containing all of the fatal alerts as determined by VADR |
vadr_docker |
String |
Docker image used to run VADR |
vadr_num_alerts |
String |
Number of fatal alerts as determined by VADR |
Titan_ClearLabs¶
The Titan_ClearLabs workflow was written to process ClearLabs WGS read data for SARS-CoV-2 Artic V3 amplicon sequencing.
Upon initiating a Titan_ClearLabs run, input ClearLabs read data provided for each sample will be processed to perform consensus genome assembly, infer the quality of both raw read data and the generated consensus genome, and assign samples SARS-CoV-2 lineage and clade types as outlined in the Titan_ClearLabs data workflow below.
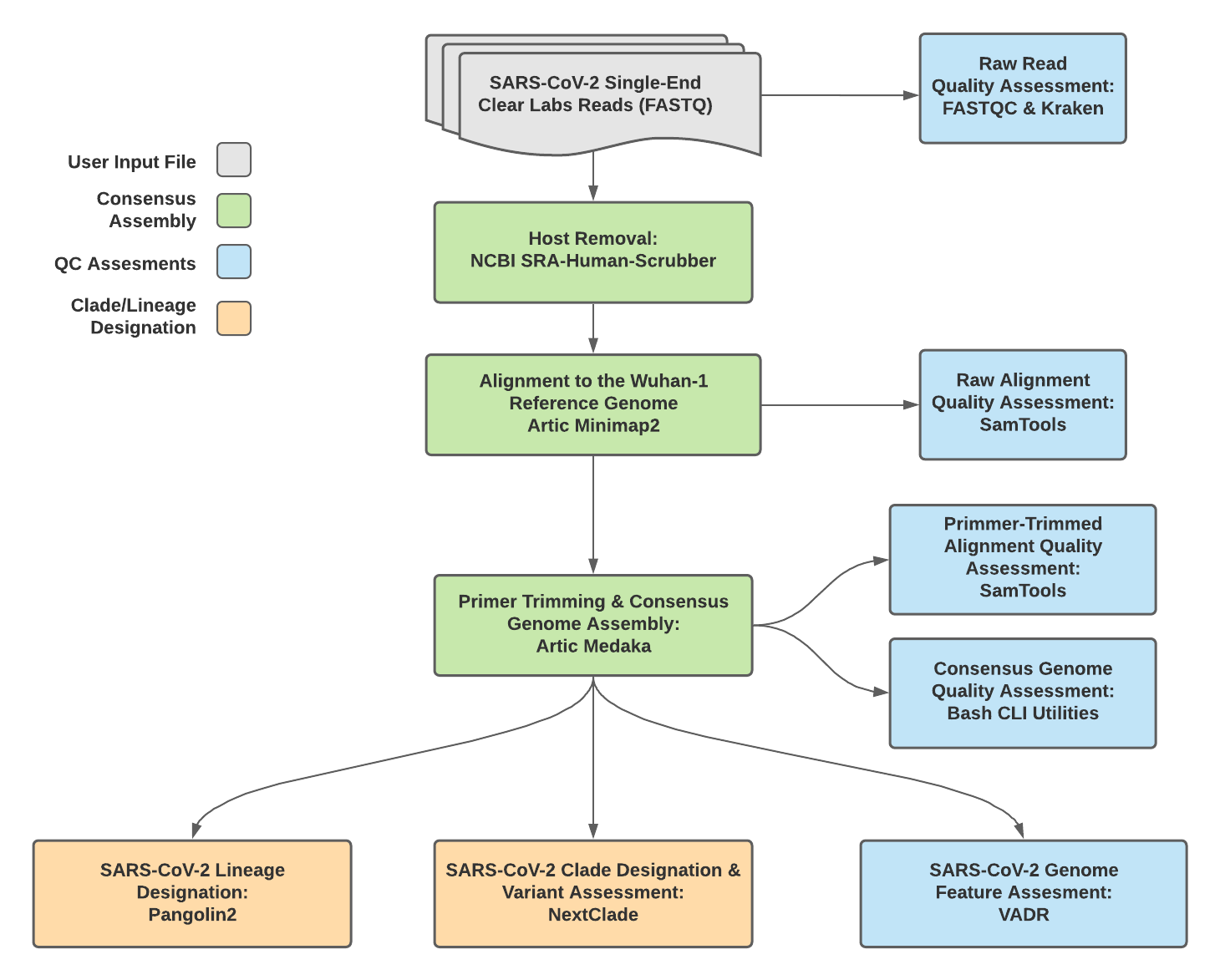
Titan_ClearLabs v1.4.4 Data Workflow¶
Consensus genome assembly with the Titan_ClearLabs workflow is performed by first de-hosting read data with the NCBI SRA-Human-Scrubber tool then following the Artic nCoV-2019 novel coronavirs bioinformatics protocol <https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html>. Briefly, input reads are aligned to the Wuhan-1 reference genome with minimap2 to generate a Binary Alignment Mapping (BAM) file. Primer sequences are then removed from the BAM file and a consensus assembly file is generated using the Artic medaka command. This assembly is then used to assign lineage and clade designations with Pangolin and NextClade. NCBI’S VADR tool is also employed to screen for potentially errant features (e.g. erroneous frame-shift mutations) in the consensus assembly.
Note
Read-trimming is performed on raw read data generated on the ClearLabs instrument and thus not a required step in the Titan_ClearLabs workflow.
More information on required user inputs, optional user inputs, default tool parameters and the outputs generated by Titan_CLearLabs are outlined below.
Required User Inputs¶
Download CSV: Titan_ClearLabs_required_inputs.csv
Task |
Input Variable |
Data Type |
Description |
|---|---|---|---|
titan_clearlabs |
clear_lab_fastq |
File |
Clear Labs FASTQ read files |
titan_clearlabs |
samplename |
String |
Name of the sample being analyzed |
Optional User Inputs¶
Download CSV: Titan_ClearLabs_optional_inputs.csv
Task |
Variable Name |
Data Type |
Description |
Default |
|---|---|---|---|---|
bedtools_cov |
primer_bed |
String |
Path to the primer sequence coordinates of the PCR scheme utilized in BED file format |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019_amplicon.bed |
bedtools_cov |
fail_threshold |
String |
Minimum coverage threshold to determin amplicon sequencing failture |
20x |
consensus |
cpu |
Int |
CPU resources allocated to the Artric Medaka task runtime environment |
8 |
fastqc_se_raw |
cpus |
Int |
CPU resources allocated to the FastQC task runtime environment for asessing raw read data |
|
fastqc_se_raw |
read1_name |
String |
Name of the sample being analyzed |
Inferred from the input read file |
kraken2_raw |
cpus |
Int |
CPU resources allocated to the Kraken task runtime environment for asessing raw read data |
4 |
kraken2_raw |
kraken2_db |
String |
Path to the reference genome within the staphb/kraken2:2.0.8-beta_hv Docker container |
/kraken2-db |
kraken2_raw |
read2 |
File |
Optional input file for the Kraken task that is not applicable to this workflow |
None |
nextclade_one_sample |
root_sequence |
File |
Custom reference sequence file for NextClade |
None |
nextclade_one_sample |
qc_config_json |
File |
Custom QC configuraiton file for NextClade |
None |
nextclade_one_sample |
pcr_primers_csv |
File |
Custom PCR primers file for NextClade |
None |
nextclade_one_sample |
gene_annotations_json |
File |
Custom gene annotation file for NextClade |
None |
nextclade_one_sample |
docker |
String |
Docker tag used for running NextClade |
neherlab/nextclade:0.14.2 |
nextclade_one_sample |
auspice_reference_tree_json |
File |
Custom reference tree file for NextClade |
None |
pangolin3 |
inference_engine |
String |
pangolin inference engine for lineage designations (usher or pangolarn) |
usher |
pangolin3 |
min_length |
Int |
Minimum query length allowed for pangolin to attempt assignment |
10000 |
pangolin3 |
max_ambig |
Float |
Maximum proportion of Ns allowed for pangolin to attempt assignment |
0.5 |
titan_clearlabs |
artic_primer_version |
String |
Version of the Artic PCR protocol used to generate input read data |
V3 |
titan_clearlabs |
normalise |
Int |
Value to normalize read counts |
200 |
titan_clearlabs |
seq_method |
String |
Description of the sequencing methodology used to generate the input read data |
ONT via Clear Labs WGS |
titan_clearlabs |
pangolin_docker_image |
String |
Docker tag used for running Pangolin |
staphb/pangolin:2.4.2-pangolearn-2021-05-19 |
vadr |
docker |
String |
Docker tag used for running VADR |
staphb/vadr:1.2.1 |
vadr |
maxlen |
Int |
Maximum length for the fasta-trim-terminal-ambigs.pl VADR script |
30000 |
vadr |
minlen |
Int |
Minimum length subsequence to possibly replace Ns for the fasta-trim-terminal-ambigs.pl VADR script |
50 |
vadr |
vadr_opts |
String |
Options for the v-annotate.pl VADR script |
–glsearch -s -r –nomisc –mkey sarscov2 –alt_fail lowscore,fstukcnf,insertnn,deletinn –mdir /opt/vadr/vadr-models/ |
vadr |
skip_length |
Int |
Minimum assembly length (unambiguous) to run vadr |
10000 |
version_capture |
timezone |
String |
User time zone in valid Unix TZ string (e.g. America/New_York) |
None |
Outputs¶
Download CSV: Titan_ClearLabs_default_outputs.csv
Output Name |
Data Type |
Description |
|---|---|---|
aligned_bai |
File |
Index companion file to the bam file generated during the consensus assembly process |
aligned_bam |
File |
Primer-trimmed BAM file; generated during conensus assembly process |
artic_version |
String |
Version of the Artic software utilized for read trimming and conesnsus genome assembly |
assembly_fasta |
File |
Consensus genome assembly |
assembly_length_unambiguous |
Int |
Number of unambiguous basecalls within the SC2 consensus assembly |
assembly_mean_coverage |
Float |
Mean sequencing depth throughout the conesnsus assembly generated after performing primer trimming–calculated using the SAMtools coverage command |
assembly_method |
String |
Method employed to generate consensus assembly |
auspice_json |
File |
Auspice-compatable JSON output generated from NextClade analysis that includes the NextClade default samples for clade-typing and the single sample placed on this tree |
consensus_flagstat |
File |
Output from the SAMtools flagstat command to assess quality of the alignment file (BAM) |
consensus_stats |
File |
Output from the SAMtools stats command to assess quality of the alignment file (BAM) |
dehosted_reads |
File |
Dehosted reads; suggested read file for SRA submission |
fastqc_clean |
Int |
Number of reads after dehosting as determined by FastQC |
fastqc_raw |
Int |
Number of raw input reads as determined by FastQC |
fastqc_version |
String |
Version of the FastQC version used |
kraken_human |
Float |
Percent of human read data detected using the Kraken2 software |
kraken_human_dehosted |
Float |
Percent of human read data detected using the Kraken2 software after host removal |
kraken_report |
String |
Full Kraken report |
kraken_report_dehosted |
File |
Full Kraken report after host removal |
kraken_sc2 |
Float |
Percent of SARS-CoV-2 read data detected using the Kraken2 software |
kraken_sc2_dehosted |
Float |
Percent of SARS-CoV-2 read data detected using the Kraken2 software after host removal |
kraken_version |
String |
Version of Kraken software used |
meanbaseq_trim |
Float |
Mean quality of the nucleotide basecalls aligned to the reference genome after primer trimming |
meanmapq_trim |
Float |
Mean quality of the mapped reads to the reference genome after primer trimming |
nextclade_aa_dels |
String |
Amino-acid deletions as detected by NextClade |
nextclade_aa_subs |
String |
Amino-acid substitutions as detected by NextClade |
nextclade_clade |
String |
NextClade clade designation |
nextclade_json |
File |
NexClade output in JSON file format |
nextclade_tsv |
File |
NextClade output in TSV file format |
nextclade_version |
String |
Version of NextClade software used |
number_Degenerate |
Int |
Number of degenerate basecalls within the consensus assembly |
number_N |
Int |
Number of fully ambiguous basecalls within the consensus assembly |
number_Total |
Int |
Total number of nucleotides within the consensus assembly |
pango_lineage |
String |
Pango lineage as detremined by Pangolin |
pango_lineage_report |
File |
Full Pango lineage report generated by Pangolin |
pangolin_conflicts |
String |
Number of lineage conflicts as deteremed by Pangolin |
pangolin_docker |
String |
Docker image used to run Pangolin |
pangolin_notes |
String |
Lineage notes as deteremined by Pangolin |
pangolin_version |
String |
Pangolin and PangoLEARN versions used |
percent_reference_coverage |
Float |
Percent coverage of the reference genome after performing primer trimming; calculated as assembly_length_unambiguous / length of reference genome (SC2: 29,903) x 100 |
pool1_percent |
Float |
Percentage of aligned read data assocaited with the pool1 amplicons |
pool2_percent |
Float |
Percentage of aligned read data assocaited with the pool 2 amplicons |
samtools_version |
String |
Version of SAMtools used to sort and index the alignment file |
seq_platform |
String |
Description of the sequencing methodology used to generate the input read data |
titan_clearlabs_analysis_date |
String |
Date of analysis |
titan_clearlabs_version |
String |
Version of the Public Health Viral Genomics (PHVG) repository used |
vadr_alerts_list |
File |
File containing all of the fatal alerts as determined by VADR |
vadr_docker |
String |
Docker image used to run VADR |
vadr_num_alerts |
String |
Number of fatal alerts as determined by VADR |
variants_from_ref_vcf |
File |
Number of variants relative to the reference genome |
Titan_ONT¶
The Titan_ONT workflow was written to process basecalled and demultiplexed Oxford Nanopore Technology (ONT) read data. IInput reads are assumed to be the product of sequencing Artic V3 tiled PCR-amplicons designed for the SARS-CoV-2 genome.
Note
As of May 2021, alternative primer schemes are not currently supported for the Titan_ONT workflow, but active development us underway to allow for such analysis in the near future.
Upon initiating a Titan_ONT run, input ONT read data provided for each sample will be processed to perform consensus genome assembly, infer the quality of both raw read data and the generated consensus genome, and assign samples SARS-CoV-2 lineage and clade types as outlined in the Titan_ONT data workflow below.
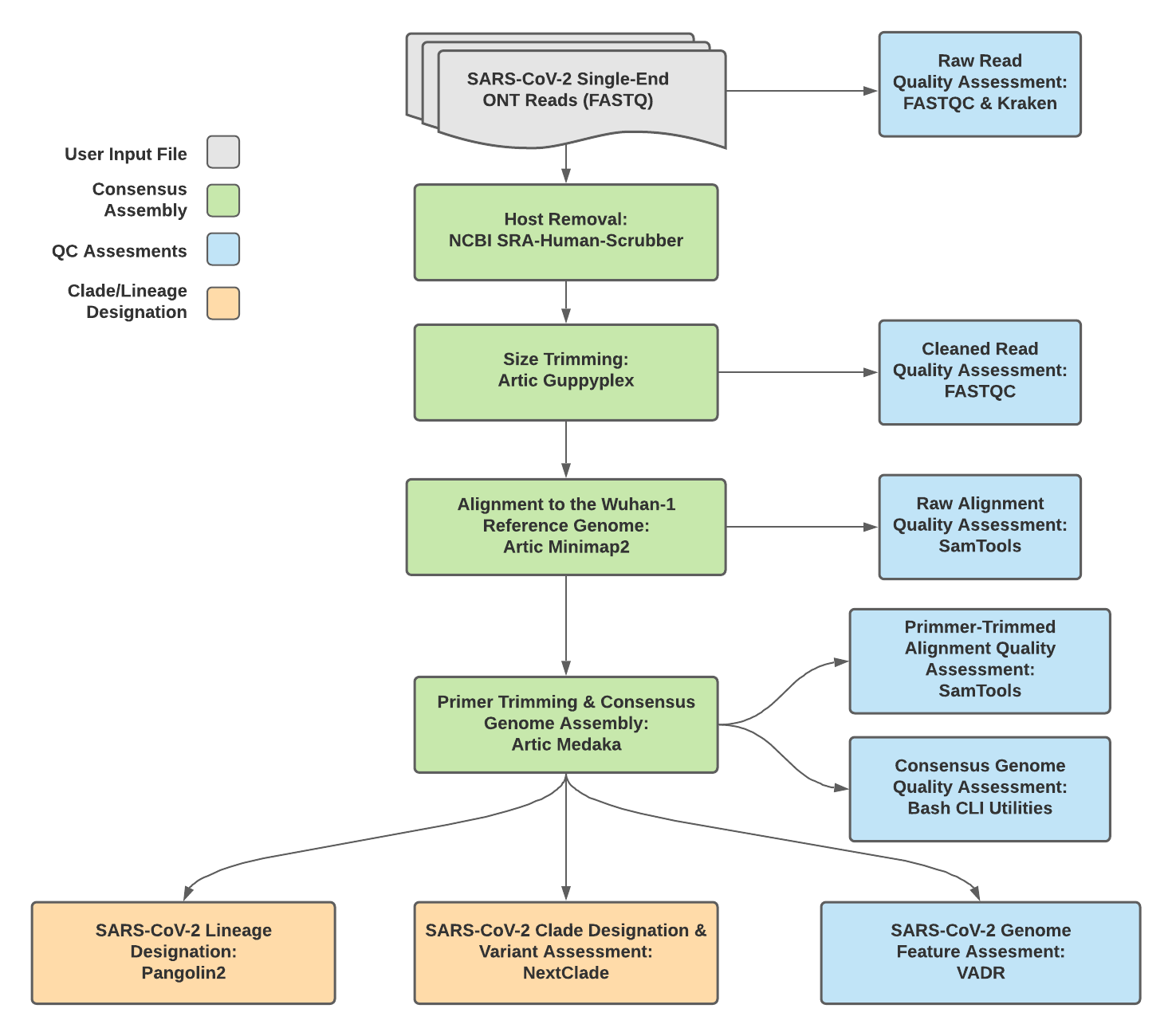
Titan_ONT v1.4.4 Data Workflow¶
Consensus genome assembly with the Titan_ONT workflow is performed performed by first de-hosting read data with the NCBI SRA-Human-Scrubber tool then following then following Artic nCoV-2019 novel coronavirs bioinformatics protocol <https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html>. Briefly, input reads are filtered by size (min-length: 400bp; max-length: 700bp) with the Aritc guppyplex command. These size-selected read data are aligned to the Wuhan-1 reference genome with minimap2 to generate a Binary Alignment Mapping (BAM) file. Primer sequences are then removed from the BAM file and a consensus assembly file is generated using the Artic medaka command. This assembly is then used to assign lineage and clade designations with Pangolin and NextClade. NCBI’S VADR tool is also employed to screen for potentially errant features (e.g. erroneous frame-shift mutations) in the consensus assembly.
More information on required user inputs, optional user inputs, default tool parameters and the outputs generated by Titan_ONT are outlined below.
Required User Inputs¶
Download CSV: Titan_ONT_required_inputs.csv
Task |
Input Variable |
Data Type |
Description |
|---|---|---|---|
titan_ont |
demultiplexed_reads |
File |
Basecalled and demultiplexed ONT read data (single FASTQ file per sample) |
titan_ont |
samplename |
String |
Name of the sample being analyzed |
Optional User Inputs¶
Download CSV: Titan_ONT_optional_inputs.csv
Task |
Variable Name |
Data Type |
Description |
Default |
|---|---|---|---|---|
bedtools_cov |
primer_bed |
String |
Path to the primer sequence coordinates of the PCR scheme utilized in BED file format |
/artic-ncov2019/primer_schemes/nCoV-2019/V3/nCoV-2019_amplicon.bed |
bedtools_cov |
fail_threshold |
String |
Minimum coverage threshold to determin amplicon sequencing failture |
20x |
consensus |
cpu |
Int |
CPU resources allocated to the Artric Medaka task runtime environment |
8 |
fastqc_se_clean |
cpus |
Int |
CPU resources allocated to the FastQC task runtime environment for asessing size-selected read data |
2 |
fastqc_se_clean |
read1_name |
String |
Name of the sample being analyzed |
Inferred from the input read file |
fastqc_se_raw |
cpus |
Int |
CPU resources allocated to the FastQC task runtime environment for asessing raw read data |
|
fastqc_se_raw |
read1_name |
String |
Name of the sample being analyzed |
Inferred from the input read file |
kraken2_raw |
cpus |
Int |
CPU resources allocated to the Kraken task runtime environment for asessing raw read data |
4 |
kraken2_raw |
kraken2_db |
String |
Path to the reference genome within the staphb/kraken2:2.0.8-beta_hv Docker container |
/kraken2-db |
kraken2_raw |
read2 |
File |
Optional input file for the Kraken task that is not applicable to this workflow |
None |
nextclade_one_sample |
root_sequence |
File |
Custom reference sequence file for NextClade |
None |
nextclade_one_sample |
qc_config_json |
File |
Custom QC configuraiton file for NextClade |
None |
nextclade_one_sample |
pcr_primers_csv |
File |
Custom PCR primers file for NextClade |
None |
nextclade_one_sample |
gene_annotations_json |
File |
Custom gene annotation file for NextClade |
None |
nextclade_one_sample |
docker |
String |
Docker tag used for running NextClade |
neherlab/nextclade:0.14.2 |
nextclade_one_sample |
auspice_reference_tree_json |
File |
Custom reference tree file for NextClade |
None |
pangolin3 |
inference_engine |
String |
pangolin inference engine for lineage designations (usher or pangolarn) |
usher |
pangolin3 |
min_length |
Int |
Minimum query length allowed for pangolin to attempt assignment |
10000 |
pangolin3 |
max_ambig |
Float |
Maximum proportion of Ns allowed for pangolin to attempt assignment |
0.5 |
read_filtering |
cpu |
Int |
CPU resources allocated to the read filtering task (Artic guppypled) runtime environment |
8 |
read_filtering |
max_length |
Int |
Maximum sequence length |
700 |
read_filtering |
min_length |
Int |
Minimum sequence length |
400 |
read_filtering |
run_prefix |
String |
Run name |
artic_ncov2019 |
titan_ont |
artic_primer_version |
String |
Version of the Artic PCR protocol used to generate input read data |
V3 |
titan_ont |
normalise |
Int |
Value to normalize read counts |
200 |
titan_ont |
seq_method |
String |
Description of the sequencing methodology used to generate the input read data |
ONT |
titan_ont |
pangolin_docker_image |
String |
Docker tag used for running Pangolin |
staphb/pangolin:2.4.2-pangolearn-2021-05-19 |
vadr |
docker |
String |
Docker tag used for running VADR |
staphb/vadr:1.2.1 |
vadr |
maxlen |
Int |
Maximum length for the fasta-trim-terminal-ambigs.pl VADR script |
30000 |
vadr |
minlen |
Int |
Minimum length subsequence to possibly replace Ns for the fasta-trim-terminal-ambigs.pl VADR script |
50 |
vadr |
vadr_opts |
String |
Options for the v-annotate.pl VADR script |
–glsearch -s -r –nomisc –mkey sarscov2 –alt_fail lowscore,fstukcnf,insertnn,deletinn –mdir /opt/vadr/vadr-models/ |
vadr |
skip_length |
Int |
Minimum assembly length (unambiguous) to run vadr |
10000 |
version_capture |
timezone |
String |
User time zone in valid Unix TZ string (e.g. America/New_York) |
None |
Outputs¶
Download CSV: Titan_ONT_default_outputs.csv
Output Name |
Data Type |
Description |
|---|---|---|
aligned_bai |
File |
Index companion file to the bam file generated during the consensus assembly process |
aligned_bam |
File |
Primer-trimmed BAM file; generated during conensus assembly process |
amp_coverage |
File |
Sequence coverage per amplicon |
artic_version |
String |
Version of the Artic software utilized for read trimming and conesnsus genome assembly |
assembly_fasta |
File |
Consensus genome assembly |
assembly_length_unambiguous |
Int |
Number of unambiguous basecalls within the SC2 consensus assembly |
assembly_mean_coverage |
Float |
Mean sequencing depth throughout the conesnsus assembly generated after performing primer trimming–calculated using the SAMtools coverage command |
assembly_method |
String |
Method employed to generate consensus assembly |
auspice_json |
File |
Auspice-compatable JSON output generated from NextClade analysis that includes the NextClade default samples for clade-typing and the single sample placed on this tree |
bedtools_version |
String |
bedtools version utilized when calculating amplicon read coverage |
consensus_flagstat |
File |
Output from the SAMtools flagstat command to assess quality of the alignment file (BAM) |
consensus_stats |
File |
Output from the SAMtools stats command to assess quality of the alignment file (BAM) |
dehosted_reads |
File |
Dehosted reads; suggested read file for SRA submission |
fastqc_clean |
Int |
Number of reads after size filttering and dehosting as determined by FastQC |
fastqc_raw |
Int |
Number of raw reads input reads as determined by FastQC |
fastqc_version |
String |
Version of the FastQC version used |
kraken_human |
Float |
Percent of human read data detected using the Kraken2 software |
kraken_human_dehosted |
Float |
Percent of human read data detected using the Kraken2 software after host removal |
kraken_report |
File |
Full Kraken report |
kraken_report_dehosted |
File |
Full Kraken report after host removal |
kraken_sc2 |
Float |
Percent of SARS-CoV-2 read data detected using the Kraken2 software |
kraken_sc2_dehosted |
Float |
Percent of SARS-CoV-2 read data detected using the Kraken2 software after host removal |
kraken_version |
String |
Version of Kraken software used |
meanbaseq_trim |
Float |
Mean quality of the nucleotide basecalls aligned to the reference genome after primer trimming |
meanmapq_trim |
Float |
Mean quality of the mapped reads to the reference genome after primer trimming |
nextclade_aa_dels |
String |
Amino-acid deletions as detected by NextClade |
nextclade_aa_subs |
String |
Amino-acid substitutions as detected by NextClade |
nextclade_clade |
String |
NextClade clade designation |
nextclade_json |
File |
NexClade output in JSON file format |
nextclade_tsv |
File |
NextClade output in TSV file format |
nextclade_version |
String |
Version of NextClade software used |
number_Degenerate |
Int |
Number of degenerate basecalls within the consensus assembly |
number_N |
Int |
Number of fully ambiguous basecalls within the consensus assembly |
number_Total |
Int |
Total number of nucleotides within the consensus assembly |
pango_lineage |
String |
Pango lineage as detremined by Pangolin |
pango_lineage_report |
File |
Full Pango lineage report generated by Pangolin |
pangolin_conflicts |
String |
Number of lineage conflicts as deteremed by Pangolin |
pangolin_docker |
String |
Docker image used to run Pangolin |
pangolin_notes |
String |
Lineage notes as deteremined by Pangolin |
pangolin_version |
String |
Pangolin and PangoLEARN versions used |
percent_reference_coverage |
Float |
Percent coverage of the reference genome after performing primer trimming; calculated as assembly_length_unambiguous / length of reference genome (SC2: 29,903) x 100 |
pool1_percent |
Float |
Percentage of aligned read data assocaited with the pool1 amplicons |
pool2_percent |
Float |
Percentage of aligned read data assocaited with the pool 2 amplicons |
samtools_version |
String |
Version of SAMtools used to sort and index the alignment file |
seq_platform |
String |
Description of the sequencing methodology used to generate the input read data |
titan_ont_analysis_date |
String |
Date of analysis |
titan_ont_version |
String |
Version of the Public Health Viral Genomics (PHVG) repository used |
vadr_alerts_list |
File |
File containing all of the fatal alerts as determined by VADR |
vadr_docker |
String |
Docker image used to run VADR |
vadr_num_alerts |
String |
Number of fatal alerts as determined by VADR |
variants_from_ref_vcf |
File |
Number of variants relative to the reference genome |