Mercury Workflow Series¶
The Mercury workflow series was developed to allow users to efficiently and accurately prepare submission files for GISAID, SRA, and Genbank submissions as well as BioSample registration. As of today (November 11th, 2021) these workflows are specific to SARS-CoV-2 amplicon read data from clinical samples, but work is underway to allow for the submission preparation of other viral pathogens of concern.
These workflows were written to ingest and properly format all suggested metadata fields as per the Public Health Alliance for Genomic Epidemiology’s SARS-CoV-2 Contextual Data Specifications.
Mercury Workflows for Single-Sample Preparation¶
Sharing of sample read and assembly data through internationally accessible databases allows insights to be drawn about how the virus is spreading and mutating across the globe; the more freely available these data are to international researchers and public health scientists, the stronger our decision making can be.
The Mercury workflows for single-sample preparation is made up of two separate WDL workflows, Mercury_SE_Prep & Mercury_PE_Prep, for preparing submission files to GISAID, SRA, and GenBank for single and paired-end read data, respectively. These two workflows will process read data, assembly files, and contextual metadata to prepare submission for samples individually–while these workflows can process multiple samples in a single run, the submission files prepared are for single-sample submission; for preparation of multiple samples (i.e. batch submission), please see details for the Mercury_Batch workflow below.
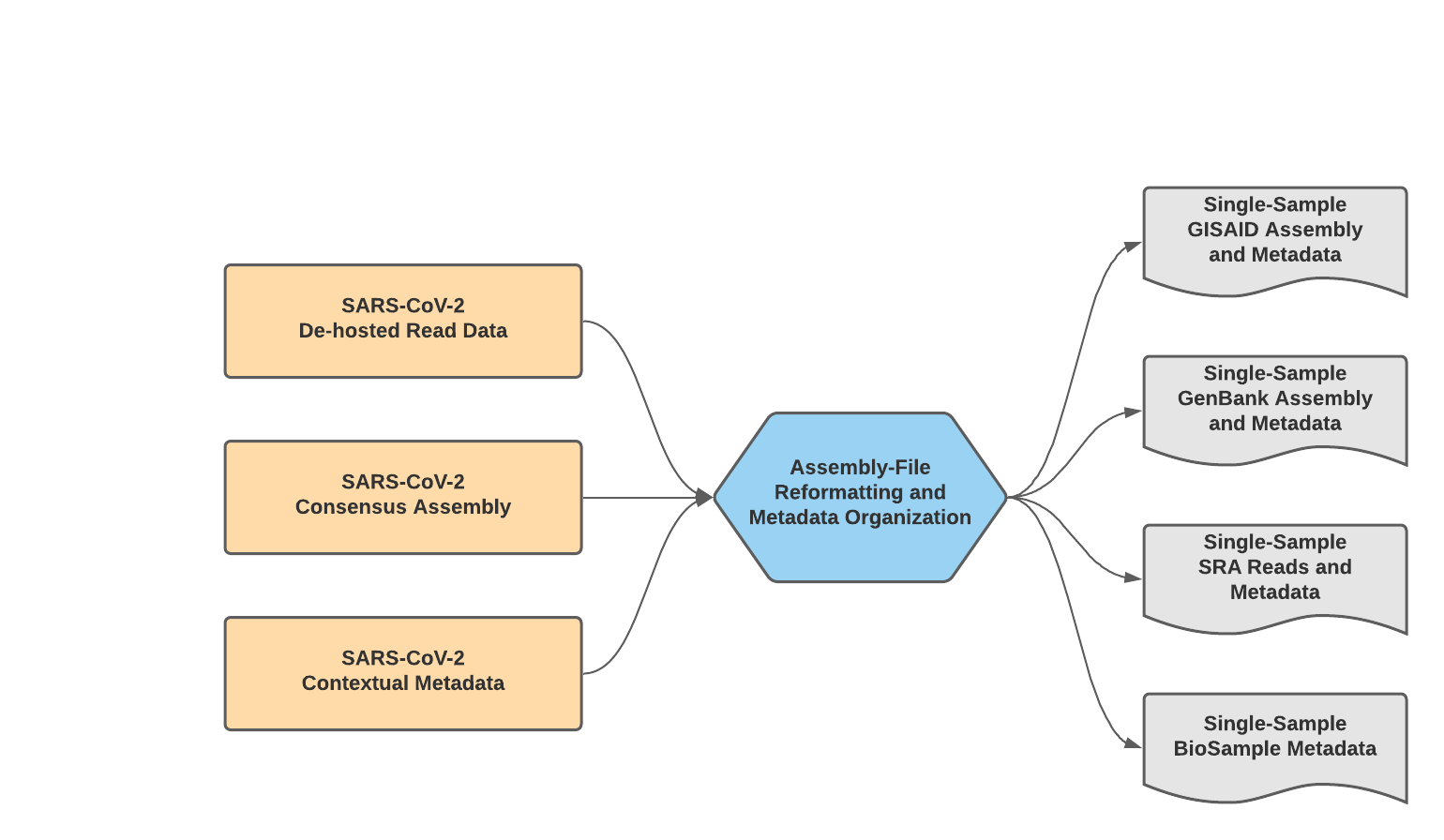
Mercury_Prep Data Workflow¶
A series of introductory training videos that provide conceptual overviews of methodologies and walkthrough tutorials on how to utilize these Mercury workflows through Terra are available on the Theiagen Genomics YouTube page:
Mercury_PE_Prep¶
The Mercury_PE_Prep workflow was written to process paired-end read data, assembly files, and contextual metadata to prepare submission for samples individually.
Note
With default settings, this workflow will only prepare submission files for samples with assembly files containing less than 5,000 Ns. This quality threshold can be adjusted by modifying the number_N_threshold.
A step-by-step video tutorial for utilizing the Mercury_PE_Prep workflow has been made available on the Theiagen YouTube Page:
More information on required user inputs, optional user inputs, default tool parameters and the outputs generated by Mercury_PE_Prep are outlined below.
Required User Inputs¶
Download CSV: Mercury_PE_Prep_required_inputs.csv
Task |
Input Variable |
Data Type |
Description |
|---|---|---|---|
mercury_pe_prep |
assembly_fasta |
File |
Consensus genome assembly |
mercury_pe_prep |
assembly_mean_coverage |
Float |
Mean sequencing depth throughout the conesnsus assembly |
mercury_pe_prep |
assembly_method |
String |
Method employed to generate the input assembbly file |
mercury_pe_prep |
authors |
String |
Authors associated with this submission |
mercury_pe_prep |
bioproject_accession |
String |
NCBI BioProject accession number |
mercury_pe_prep |
collecting_lab |
String |
Name of the laboratory that orginial laboratory that collected the sample |
mercury_pe_prep |
collecting_lab_address |
String |
Address of the laboratory that orginial laboratory that collected the sample |
mercury_pe_prep |
collection_date |
String |
Date on which the sample was collected |
mercury_pe_prep |
continent |
String |
Continent the sample was collected in |
mercury_pe_prep |
country |
String |
Country the sample was collected in |
mercury_pe_prep |
gisaid_submitter |
String |
GISAID username |
mercury_pe_prep |
host_disease |
String |
Host disease; for SARS-CoV-2 sequences from human samples, “COVID-19” would be the most accurate entry for this field |
mercury_pe_prep |
instrument_model |
String |
Model of the sequencing instrument utilized to generate the read data |
mercury_pe_prep |
isolation_source |
String |
Isolation source, i.e. clinical, animal, or environmental |
mercury_pe_prep |
library_id |
String |
Unique identifer for the sequenced library |
mercury_pe_prep |
library_selection |
String |
Selection methodology used to designate samples as eligible for sequencing, e.g., “PCR” for samples selected based on PCT Ct values |
mercury_pe_prep |
library_source |
String |
Source of the genomic material used to prepare the sequencing libraries |
mercury_pe_prep |
library_strategy |
String |
Library preparation strategy, e.g., “AMPLICON” for data generated from tiling PCR amplicons |
mercury_pe_prep |
number_N |
Int |
Number of fully ambiguous basecalls within the consensus assembly |
mercury_pe_prep |
organism |
String |
Name of the organism sequenced, e.g. “SARS-CoV-2” |
mercury_pe_prep |
read1_dehosted |
File |
Dehosted forward read file |
mercury_pe_prep |
read2_dehosted |
File |
Dehosted reverse read file |
mercury_pe_prep |
seq_platform |
String |
Description of the sequencing methodology used to generate the input read data |
mercury_pe_prep |
state |
String |
State the sample was collected in |
mercury_pe_prep |
submission_id |
String |
Unique identfier for the sample utilized upon submission |
mercury_pe_prep |
submitting_lab |
String |
Name of the submitting laboratory |
mercury_pe_prep |
submitting_lab_address |
String |
Address of the submitting laboratory |
Optional User Inputs¶
Download CSV: Mercury_PE_Prep_optional_inputs.csv
Task |
Input Variable |
Data Type |
Description |
Default |
|---|---|---|---|---|
gisaid_prep_one_sample |
specimen_source |
String |
Biologial source of the specimen, e.g. e.g. sputum, Alveolar lavage fluid, Oro-pharyngeal swab, Blood, Tracheal swab, Urine, Stool, Cloakal swab, Organ, Feces, Other |
None |
gisaid_prep_one_sample |
mem_size_gb |
Int |
Memory allocated to the gisaid_prep_one_sample task |
1 |
gisaid_prep_one_sample |
disk_size |
Int |
Disk size allocated to the gisaid_prep_one_sample task |
25 |
gisaid_prep_one_sample |
patient_status |
String |
Status of the patient, e.g. Hospitalized, Released, Live, Deceased, unknown |
unknown |
gisaid_prep_one_sample |
type |
String |
Organism typoe |
betacoronovirus |
gisaid_prep_one_sample |
CPUs |
Int |
CPUs allocated to the gisaid_prep_one_sample task |
None |
gisaid_prep_one_sample |
preemptible_tries |
Int |
Number of preemptible tries for the gisaid_prep_one_sample task |
0 |
gisaid_prep_one_sample |
outbreak |
String |
Outbreak associated with this submision, e.g. date, place, family cluster |
None |
gisaid_prep_one_sample |
last_vaccinated |
String |
Date of last vaccine recieved |
None |
gisaid_prep_one_sample |
docker_image |
String |
Docker image utilized for the gisaid_prep_one_sample task |
quay.io/theiagen/utility:1.1 |
gisaid_prep_one_sample |
passage_details |
String |
Passage details of the sample being submitted, e.g. original, vero, etc |
original |
mercury_pe_prep |
dehosting_method |
String |
Method utilized to dehost read data |
NCBI Human Scrubber |
mercury_pe_prep |
filetype |
String |
File type of the read data being submitted to SRA |
fastq |
mercury_pe_prep |
submitter_email |
String |
Email address of the submitter |
None |
mercury_pe_prep |
purpose_of_sequencing |
String |
Reason that this sample was sequenced; for labs that are sequencing samples as part of a federal surveillance program “baseline surveillance” would be the most accurate entry for this field |
None |
mercury_pe_prep |
library_layout |
String |
Layout of the sequenced library |
paired |
mercury_pe_prep |
number_N_threshold |
Int |
Maximum number of ambiguous nucleotides in a sample to prepare submission files |
5000 |
mercury_pe_prep |
host_sci_name |
String |
Scientific name of the host organism |
Homo sapiens |
mercury_pe_prep |
gisaid_accession |
String |
Accession number in GISAID |
None |
mercury_pe_prep |
gisaid_organism |
String |
Orgiansm name as per GISAID submission |
hCoV-19 |
mercury_pe_prep |
county |
String |
County the laboratory was collected in |
None |
mercury_pe_prep |
amplicon_size |
String |
Average size of the amplicons sequenced |
None |
mercury_pe_prep |
host |
String |
Common name of the host organism |
Human |
mercury_pe_prep |
amplicon_primer_scheme |
String |
Name of the amplicon primer scheme utilized to generate the amplicons sequenced |
None |
mercury_pe_prep |
biosample_accession |
String |
BioSample accession number |
None |
mercury_pe_prep |
treatment |
String |
Treatment administered to the patient, e.g. drug name, dosage, etc. |
None |
mercury_pe_prep |
patient_gender |
String |
Gender of the patient |
unknown |
mercury_pe_prep |
purpose_of_sampling |
String |
Reason that the original specimen was taken, e.g. clinical diagnostics |
None |
mercury_pe_prep |
patient_age |
String |
Age of the patient |
unknown |
ncbi_prep_one_sample |
mem_size_gb |
Int |
Memory allocated to the ncbi_prep_one_sample task |
1 |
ncbi_prep_one_sample |
docker_image |
String |
Docker image utilized for the ncbi_prep_one_sample task |
quay.io/staphb/vadr:1.3 |
ncbi_prep_one_sample |
maxlen |
Int |
VADR –maxlen input utilized when trimming terminal ambiguous ends |
30000 |
ncbi_prep_one_sample |
preemptible_tries |
Int |
Number of preemptible tries for the ncbi_prep_one_sample task |
0 |
ncbi_prep_one_sample |
CPUs |
Int |
CPUs allocated to the ncbi_prep_one_sample task |
1 |
ncbi_prep_one_sample |
minlen |
Int |
VADR –minen input utilized when trimming terminal ambiguous ends |
50 |
ncbi_prep_one_sample |
disk_size |
Int |
Disk size allocated the ncbi_prep_one_sample task |
25 |
version_capture |
timezone |
String |
User time zone in valid Unix TZ string (e.g. America/New_York) |
None |
Outputs¶
Download CSV: Mercury_PE_Prep_default_outputs.csv
Output Name |
Data Type |
Description |
|---|---|---|
biosample_attributes |
File |
Sample metadata compiled and formatted to meet the BioSample submission requirements |
genbank_assembly |
File |
Assembly file reformatted to meet the GenBank submission requirements |
genbank_modifier |
File |
Sample metadata compiled and formatted to meet the GenBank submission requirements; will need to be manually modified to include BioSample accession numbers |
gisaid_assembly |
File |
Assembly file reformatted to meet the GISAID submission requirements |
gisaid_metadata |
File |
Metadata compiled and formatted to meet the GISAID submission requirements |
mercury_pe_prep_analysis_date |
String |
Date of analysis |
mercury_pe_prep_version |
String |
Version of the Public Health Viral Genomics (PHVG) repository used |
sra_metadata |
File |
Sample and read metadata compiled and formatted to meet the SRA submission requirements |
sra_read1 |
File |
Forward read formatted for submission to SRA |
sra_read2 |
File |
Reverse read formatted for submission to SRA |
sra_reads |
File |
Forward and reverse reads formatted for submission to SRA |
Mercury_SE_Prep¶
The Mercury_SE_Prep workflow was written to process single-end read data, assembly files, and contextual metadata to prepare submission for samples individually.
Note
With default settings, this workflow will only prepare submission files for samples with assembly files containing less than 5,000 Ns. This quality threshold can be adjusted by modifying the number_N_threshold.
More information on required user inputs, optional user inputs, default tool parameters and the outputs generated by Mercury_SE_Prep are outlined below.
Required User Inputs¶
Download CSV: Mercury_SE_Prep_required_inputs.csv
Task |
Input Variable |
Data Type |
Description |
|---|---|---|---|
mercury_pe_prep |
assembly_fasta |
File |
Consensus genome assembly |
mercury_pe_prep |
assembly_mean_coverage |
Float |
Mean sequencing depth throughout the conesnsus assembly |
mercury_pe_prep |
assembly_method |
String |
Method employed to generate the input assembbly file |
mercury_pe_prep |
authors |
String |
Authors associated with this submission |
mercury_pe_prep |
bioproject_accession |
String |
NCBI BioProject accession number |
mercury_pe_prep |
collecting_lab |
String |
Name of the laboratory that orginial laboratory that collected the sample |
mercury_pe_prep |
collecting_lab_address |
String |
Address of the laboratory that orginial laboratory that collected the sample |
mercury_pe_prep |
collection_date |
String |
Date on which the sample was collected |
mercury_pe_prep |
continent |
String |
Continent the sample was collected in |
mercury_pe_prep |
country |
String |
Country the sample was collected in |
mercury_pe_prep |
gisaid_submitter |
String |
GISAID username |
mercury_pe_prep |
host_disease |
String |
Host disease; for SARS-CoV-2 sequences from human samples, “COVID-19” would be the most accurate entry for this field |
mercury_pe_prep |
instrument_model |
String |
Model of the sequencing instrument utilized to generate the read data |
mercury_pe_prep |
isolation_source |
String |
Isolation source, i.e. clinical, animal, or environmental |
mercury_pe_prep |
library_id |
String |
Unique identifer for the sequenced library |
mercury_pe_prep |
library_selection |
String |
Selection methodology used to designate samples as eligible for sequencing, e.g., “PCR” for samples selected based on PCT Ct values |
mercury_pe_prep |
library_source |
String |
Source of the genomic material used to prepare the sequencing libraries |
mercury_pe_prep |
library_strategy |
String |
Library preparation strategy, e.g., “AMPLICON” for data generated from tiling PCR amplicons |
mercury_pe_prep |
number_N |
Int |
Number of fully ambiguous basecalls within the consensus assembly |
mercury_pe_prep |
organism |
String |
Name of the organism sequenced, e.g. “SARS-CoV-2” |
mercury_pe_prep |
reads_dehosted |
File |
Dehosted read files |
mercury_pe_prep |
seq_platform |
String |
Description of the sequencing methodology used to generate the input read data |
mercury_pe_prep |
state |
String |
State the sample was collected in |
mercury_pe_prep |
submission_id |
String |
Unique identfier for the sample utilized upon submission |
mercury_pe_prep |
submitting_lab |
String |
Name of the submitting laboratory |
mercury_pe_prep |
submitting_lab_address |
String |
Address of the submitting laboratory |
Optional User Inputs¶
Download CSV: Mercury_SE_Prep_optional_inputs.csv
Task |
Input Variable |
Data Type |
Description |
Default |
|---|---|---|---|---|
gisaid_prep_one_sample |
specimen_source |
String |
Biologial source of the specimen, e.g. e.g. sputum, Alveolar lavage fluid, Oro-pharyngeal swab, Blood, Tracheal swab, Urine, Stool, Cloakal swab, Organ, Feces, Other |
None |
gisaid_prep_one_sample |
mem_size_gb |
Int |
Memory allocated to the gisaid_prep_one_sample task |
1 |
gisaid_prep_one_sample |
disk_size |
Int |
Disk size allocated to the gisaid_prep_one_sample task |
25 |
gisaid_prep_one_sample |
patient_status |
String |
Status of the patient, e.g. Hospitalized, Released, Live, Deceased, unknown |
unknown |
gisaid_prep_one_sample |
type |
String |
Organism typoe |
betacoronovirus |
gisaid_prep_one_sample |
CPUs |
Int |
CPUs allocated to the gisaid_prep_one_sample task |
None |
gisaid_prep_one_sample |
preemptible_tries |
Int |
Number of preemptible tries for the gisaid_prep_one_sample task |
0 |
gisaid_prep_one_sample |
outbreak |
String |
Outbreak associated with this submision, e.g. date, place, family cluster |
None |
gisaid_prep_one_sample |
last_vaccinated |
String |
Date of last vaccine recieved |
None |
gisaid_prep_one_sample |
docker_image |
String |
Docker image utilized for the gisaid_prep_one_sample task |
quay.io/theiagen/utility:1.1 |
gisaid_prep_one_sample |
passage_details |
String |
Passage details of the sample being submitted, e.g. original, vero, etc |
original |
mercury_pe_prep |
dehosting_method |
String |
Method utilized to dehost read data |
NCBI Human Scrubber |
mercury_pe_prep |
filetype |
String |
File type of the read data being submitted to SRA |
fastq |
mercury_pe_prep |
submitter_email |
String |
Email address of the submitter |
None |
mercury_pe_prep |
purpose_of_sequencing |
String |
Reason that this sample was sequenced; for labs that are sequencing samples as part of a federal surveillance program “baseline surveillance” would be the most accurate entry for this field |
None |
mercury_pe_prep |
library_layout |
String |
Layout of the sequenced library |
paired |
mercury_pe_prep |
number_N_threshold |
Int |
Maximum number of ambiguous nucleotides in a sample to prepare submission files |
5000 |
mercury_pe_prep |
host_sci_name |
String |
Scientific name of the host organism |
Homo sapiens |
mercury_pe_prep |
gisaid_accession |
String |
Accession number in GISAID |
None |
mercury_pe_prep |
gisaid_organism |
String |
Orgiansm name as per GISAID submission |
hCoV-19 |
mercury_pe_prep |
county |
String |
County the laboratory was collected in |
None |
mercury_pe_prep |
amplicon_size |
String |
Average size of the amplicons sequenced |
None |
mercury_pe_prep |
host |
String |
Common name of the host organism |
Human |
mercury_pe_prep |
amplicon_primer_scheme |
String |
Name of the amplicon primer scheme utilized to generate the amplicons sequenced |
None |
mercury_pe_prep |
biosample_accession |
String |
BioSample accession number |
None |
mercury_pe_prep |
treatment |
String |
Treatment administered to the patient, e.g. drug name, dosage, etc. |
None |
mercury_pe_prep |
patient_gender |
String |
Gender of the patient |
unknown |
mercury_pe_prep |
purpose_of_sampling |
String |
Reason that the original specimen was taken, e.g. clinical diagnostics |
None |
mercury_pe_prep |
patient_age |
String |
Age of the patient |
unknown |
ncbi_prep_one_sample |
mem_size_gb |
Int |
Memory allocated to the ncbi_prep_one_sample task |
1 |
ncbi_prep_one_sample |
docker_image |
String |
Docker image utilized for the ncbi_prep_one_sample task |
quay.io/staphb/vadr:1.3 |
ncbi_prep_one_sample |
maxlen |
Int |
VADR –maxlen input utilized when trimming terminal ambiguous ends |
30000 |
ncbi_prep_one_sample |
preemptible_tries |
Int |
Number of preemptible tries for the ncbi_prep_one_sample task |
0 |
ncbi_prep_one_sample |
CPUs |
Int |
CPUs allocated to the ncbi_prep_one_sample task |
1 |
ncbi_prep_one_sample |
minlen |
Int |
VADR –minen input utilized when trimming terminal ambiguous ends |
50 |
ncbi_prep_one_sample |
disk_size |
Int |
Disk size allocated the ncbi_prep_one_sample task |
25 |
version_capture |
timezone |
String |
User time zone in valid Unix TZ string (e.g. America/New_York) |
None |
Outputs¶
Download CSV: Mercury_SE_Prep_default_outputs.csv
Output Name |
Data Type |
Description |
|---|---|---|
biosample_attributes |
File |
Sample metadata compiled and formatted to meet the BioSample submission requirements |
genbank_assembly |
File |
Assembly file reformatted to meet the GenBank submission requirements |
genbank_modifier |
File |
Sample metadata compiled and formatted to meet the GenBank submission requirements; will need to be manually modified to include BioSample accession numbers |
gisaid_assembly |
File |
Assembly file reformatted to meet the GISAID submission requirements |
gisaid_metadata |
File |
Metadata compiled and formatted to meet the GISAID submission requirements |
mercury_pe_prep_analysis_date |
String |
Date of analysis |
mercury_pe_prep_version |
String |
Version of the Public Health Viral Genomics (PHVG) repository used |
sra_metadata |
File |
Sample and read metadata compiled and formatted to meet the SRA submission requirements |
sra_reads |
File |
Forward and reverse reads formatted for submission to SRA |
Mercury Workflows for Multiple-Sample (Batch) Preparation¶
We have made a single WDL workflow for multiple-sample (batch) preparation: Mercury_Batch.
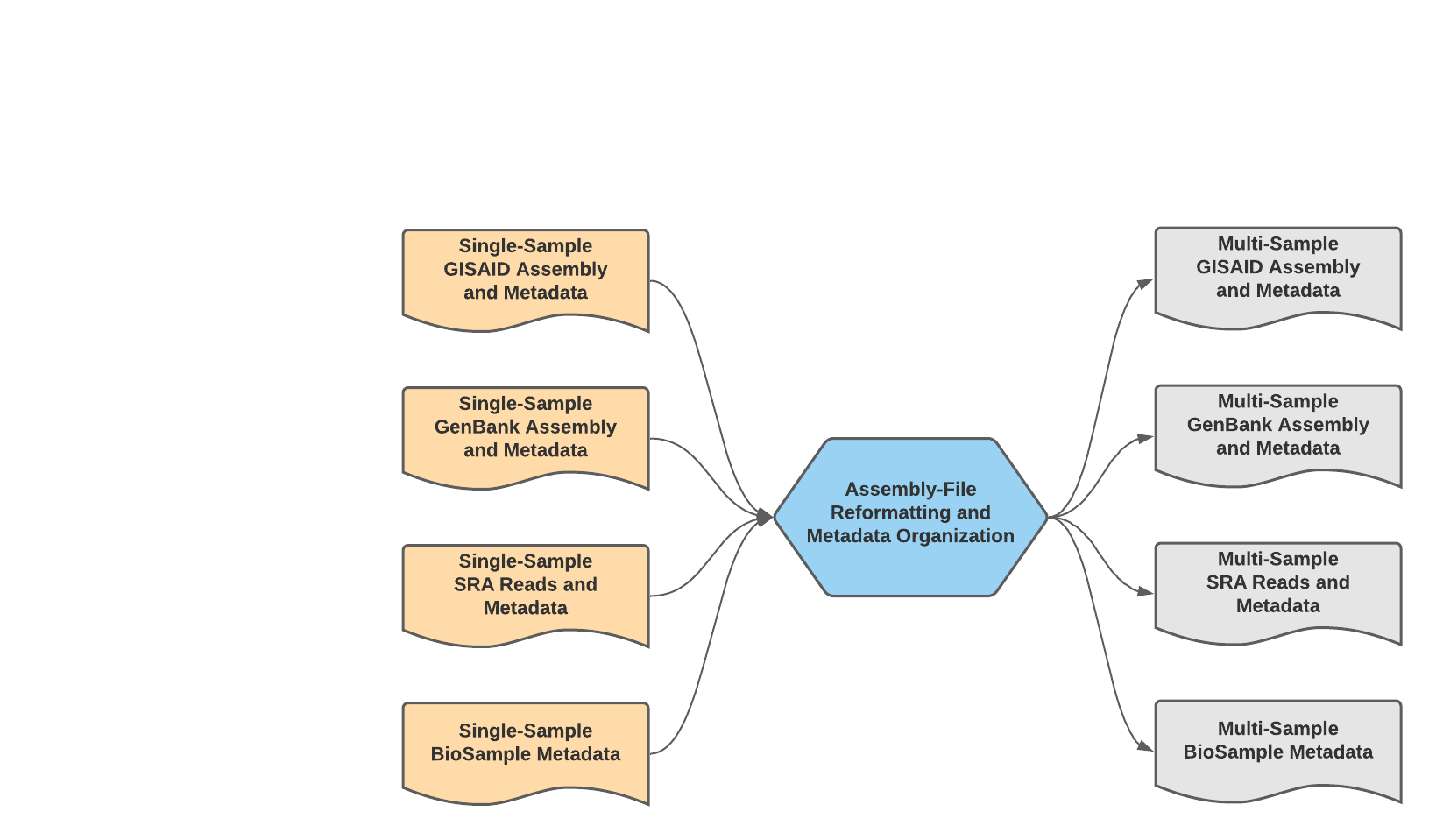
Mercury_Batch Data Workflow¶
Mercury_Batch¶
The Mercury_Batch workflow was written to process the output submission files from Mercury_PE_Prep or Mercury_SE_Prep and combine them to enable GISAID, SRA, and Genbank batch submission as well as batch BioSample registration. To avoid issues with NCBI GenBank rejections, the Mercury_Batch workflow will remove any sample with raised VADR alerts from the prepared batch submission files.
Note
With default settings, this workflow will remove samples any sample with one or more raised VADR alerts. This screening threshold can be adjusted by modifying the vadr_threshold.
A step-by-step video tutorial for utilizing the Mercury_Batch workflow has been made available on the Theiagen YouTube Page:
More information on required user inputs, optional user inputs, default tool parameters and the outputs generated by Mercury_Batch are outlined below.
Required User Inputs¶
Download CSV: Mercury_Batch_required_inputs.csv
Task |
Input Variable |
Data Type |
Description |
|---|---|---|---|
mercury_batch |
biosample_attributes |
Array[File] |
Array of sample metadata filescompiled and formatted to meet the BioSample submission requirements |
mercury_batch |
genbank_assembly |
Array[File] |
Array of assembly files reformatted to meet the GenBank submission requirements |
mercury_batch |
genbank_modifier |
Array[File] |
Array of sample metadata files compiled and formatted to meet the GenBank submission requirements; will need to be manually modified to include BioSample accession numbers |
mercury_batch |
gisaid_assembly |
Array[File] |
Array of metadata files compiled and formatted to meet the GISAID submission requirements |
mercury_batch |
gisaid_metadata |
Array[File] |
Array of assembly files reformatted to meet the GISAID submission requirements |
mercury_batch |
samplename |
Array[String] |
Array of sample identifiers |
mercury_batch |
sra_metadata |
Array[File] |
Array of sample and read metadata files compiled and formatted to meet the SRA submission requirements |
mercury_batch |
sra_reads |
Array[String] |
Array of forward and reverse reads formatted for submission to SRA |
mercury_batch |
submission_id |
Array[String] |
Array of submission identifiers |
mercury_batch |
vadr_num_alerts |
Array[String] |
Array of VADR number of alerts |
Optional User Inputs¶
Download CSV: Mercury_Batch_optional_inputs.csv
Task |
Input Variable |
Data Type |
Description |
Default |
|---|---|---|---|---|
compile_biosamp_n_sra |
docker_image |
String |
Docker image utilized for the compile_biosample_n_sra task |
quay.io/theiagen/utility:1.1 |
compile_biosamp_n_sra |
preemptible_tries |
Int |
Number of preemptible tries for the compile_biosample_n_sra task |
0 |
genbank_compile |
docker_image |
String |
Docker image utilized for the genbank_compile task |
quay.io/theiagen/utility:1.1 |
genbank_compile |
preemptible_tries |
Int |
Number of preemptible tries for the genbank_compile task |
0 |
gisaid_compile |
docker_image |
String |
Docker image utilized for the gisaid_compile task |
quay.io/theiagen/utility:1.1 |
gisaid_compile |
preemptible_tries |
Int |
Number of preemptible tries for the gisaid_compile task |
0 |
mercury_batch |
CPUs |
Int |
CPUs allocated for each task in the mercury_batch workflow |
4 |
mercury_batch |
disk_size |
Int |
Disk size allocated for each task in the mercury_batch workflow |
100 |
mercury_batch |
gcp_bucket |
String |
GCP bucket for SRA transfer |
None |
mercury_batch |
mem_size_gb |
Int |
Memory allocated for each task in the mercury_batch workflow |
8 |
mercury_batch |
vadr_threshold |
Int |
Maximum number of VADR alerts for samples included in the batch submission files |
0 |
version_capture |
timezone |
String |
User time zone in valid Unix TZ string (e.g. America/New_York) |
None |
Outputs¶
Download CSV: Mercury_Batch_default_outputs.csv
Output Name |
Data Type |
Description |
|---|---|---|
GenBank_batched_samples |
File |
File detailing all of the files bacthed for GenBank submission |
GenBank_excluded_samples |
File |
File detailing all of the files excluded from the prepared submission files for GenBank |
GenBank_modifier |
File |
Compiled matadata formatted for batch submissinon to GenBank |
GISAID_assembly |
File |
Concatenated assemly file for batch submission to GenBank |
GISAID_batched_samples |
File |
File detailing all of the files bacthed for GenBank submission |
GISAID_excluded_samples |
File |
File detailing all of the files excluded from the prepared submission files for GenBank |
GISAID_metadata |
File |
Compiled metadata formatted for batch submissino to GISAID |
mercury_batch_analysis_date |
String |
Date of analysis |
mercury_batch_version |
String |
Version of the Public Health Viral Genomics (PHVG) repository used |
SRA_gcp_bucket |
String |
GCP bucket location for SRA read transfer |
SRA_metadata |
File |
Compiled metadata formatted for batch submissino to SRA |
SRA_zipped_reads |
File |
All reads prepared for SRA submission (empty file is GCP bucket location was provided for SRA read transfer) |